Genomics: Insight
Approaches for gene-targeted therapies for rare disease: A Focus on Neurofibromatosis type 1
Research Question: How can gene-targeted therapies be developed and optimized to effectively treat Neurofibromatosis-1 (NF-1) and other rare diseases?
Introduction
Rare diseases are diseases that affect a small number of people compared to the general population. In Europe, a disease is considered rare if it affects fewer than 50 per 100,000 people, while in the United States, the threshold is fewer than 200,000 individuals, or approximately 60 per 100,000 people1. Rare diseases are often under-researched, leading to limited treatment options and a high burden on affected individuals and their families.
Neurofibromatosis type 1 (NF-1) is an autosomal dominant condition, meaning that a single copy of the mutated gene inherited from either parent can cause the disorder. NF-1 affects approximately 1 in 2,500 to 4,000 individuals worldwide2,3 The condition is caused by mutations in the NF1 gene located on chromosome 17, which encodes the protein neurofibromin. Neurofibromin acts as a tumor suppressor by regulating cell growth and preventing cells from proliferating uncontrollably3. Individuals with NF-1 typically have one normal and one mutated copy of the NF1 gene. Notably, about half of all NF-1 cases result from spontaneous mutations, meaning they occur in individuals with no family history of the disorder4,5.
Patients with NF-1 experience a wide range of symptoms, as the condition can manifest differently even among family member4,6. A common complication is the development of multiple neurofibromas, benign tumors that form along nerves in the skin, brain, and other areas4. The formation of this tumors require an additional somatic “second-hit” mutation in the NF1 gene7. Other key clinical signs include multiple café-au-lait spots, freckling in skin folds, numerous skin neurofibromas, subcutaneous or deep nodular neurofibromas, plexiform neurofibromas, and characteristic eye findings6. Additionally, individuals with NF-1 often face challenges related to learning, behavior, and social adaptation6.
Diagnosing and treating NF-1
Diagnosing NF-1 generally combines clinical evaluation with genetic testing. Clinically, NF-1 is identified by characteristic signs such as café-au-lait spots, neurofibromas, and Lisch nodules in the eyes4. Genetic testing, though not always required, can confirm the diagnosis by detecting mutations in the NF1 gene4.
Current treatments for NF-1 focus on managing symptoms, including surgical removal of tumors and addressing complications. For children aged 2 and older, the Food and Drug Administration (FDA) has approved selumetinib (Koselugo) to inhibit tumor growth. Selumetinib is a medication specifically designed to target and inhibit a pathway in cells known as the MAPK pathway, which can become overactive in individuals with NF-1 due to mutations in the NF1 gene8. By blocking the activity of certain proteins in this
pathway, selumetinib reduces the growth and size of tumors associated with NF1. This targeted therapy addresses the hyperactive signaling downstream of Ras, a key protein involved in cell growth and division, thereby helping to manage the tumor burden in patients with NF18. Surgery may also be performed to remove symptomatic tumors or correct bone malformations like scoliosis9. Chemotherapy is used to treat optic pathway gliomas and other cancers associated with NF-16,9. However, these treatments do not address the underlying genetic cause, limiting their long-term effectiveness.
A promising area for NF-1 treatment is gene-targeted therapies, which offer personalized approaches by modulating gene expression and protein function at various levels, including RNA, DNA, and protein (Figure 1). This review evaluates how these therapies could improve treatment outcomes and quality of life for NF1 patients.
Rare diseases are often under-researched, leading to limited treatment options and a high burden on affected individuals.
Gene-Targeted Therapies for NF-1
Gene-targeted therapies encompass a range of techniques designed to modify disease-causing gene variants or their products10. These therapies represent a novel class of treatments that can modulate gene expression and protein function at the RNA level, offering new options for various diseases, including cancers, infectious diseases, and neurodegenerative disorders10. While extensively researched in oncology, the potential of gene-targeted therapies for treating genetic conditions like NF1, which predispose individuals to tumors, remains less explored.
- These therapies include, but are not limited to:
- Gene replacement
- Genome editing with and without CRISPR-Cas9
- RNA editing
- Transcription, translation, and ubiquitination modulators (small molecules and ASOs)
- Nonsense suppression therapy (NST)
Each of these approaches has unique mechanisms of action and potential applications in treating genetic disorders like NF-1.
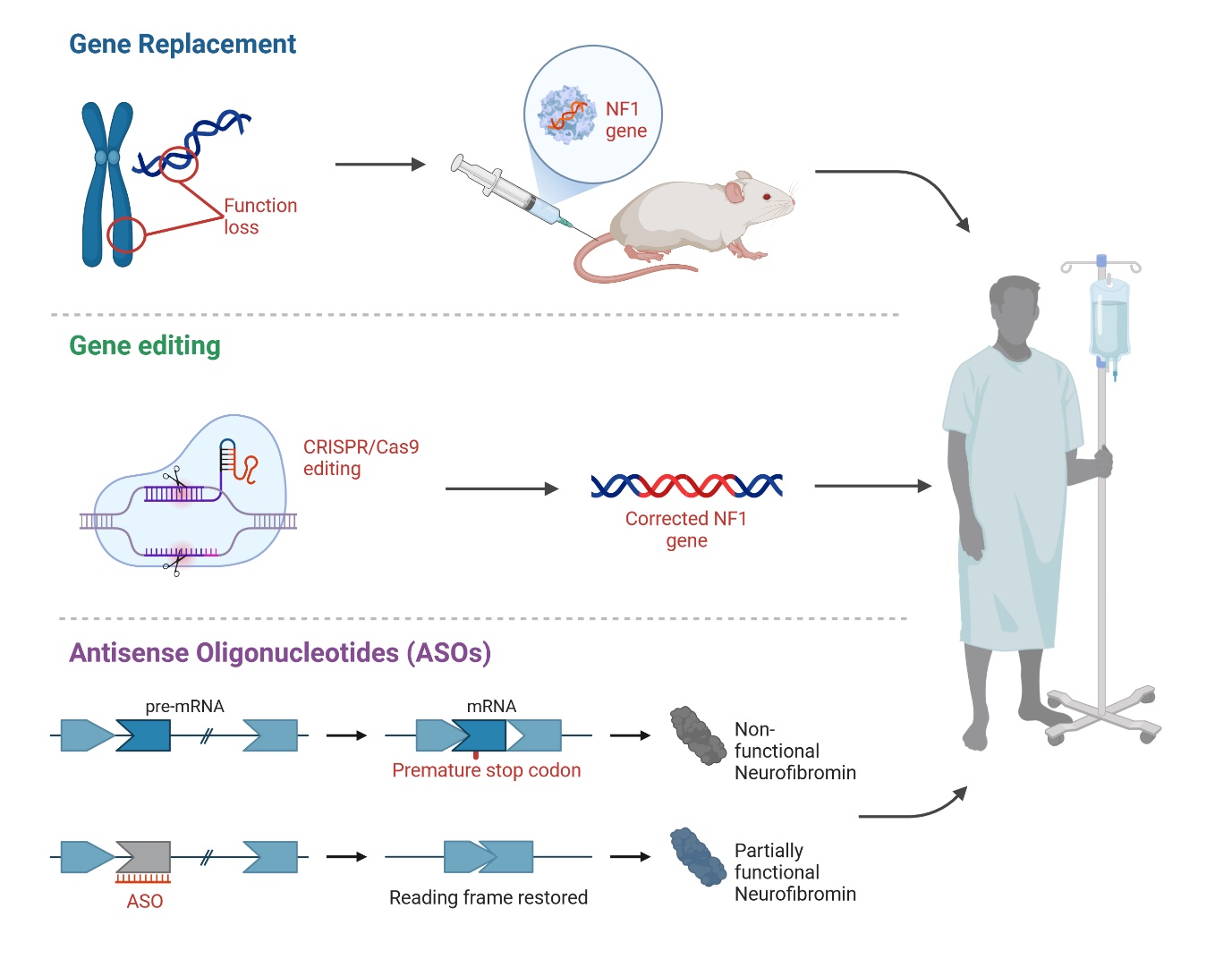
Gene Replacement: Gene therapy aims to add a functional copy of a gene to cells with defective ones, as seen with Luxturna, the first FDA-approved gene replacement therapy, which delivers a normal RPE65 gene to retinal cells to help restore vision loss11. However, it’s not a cure, and improvements may not last. Treating NF-1 with gene replacement is difficult because of the large size and complexity of the NF1 gene[10,11]. A preclinical study showed that AAV-mediated delivery of the NF1 gene to Schwann cells in a mouse model resulted in a significant reduction in tumor burden and improved survival compared to untreated mice (Figure 1)12. A new approach is being tested that uses nanoparticles to deliver the full NF1 gene, which could potentially address a wide range of mutations11.
Gene editing: Gene editing offers an alternative to whole-gene replacement by directly repairing small DNA or RNA mutations. Major DNA repair systems include zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and CRISPR-Cas proteins, with CRISPR-Cas9 showing particular promise[11] This system uses a guide RNA to target specific DNA sequences and a Cas9 enzyme to cut and modify
the DNA11. The potential of CRISPR/Cas9 technology in treating genetic diseases, including NF-1, has generated considerable excitement13,14. One notable effort is the Gilbert Family Foundation's Gene Therapy Initiative, which includes projects aimed at developing CRISPR-based genome editing tools to correct NF1 mutations. These projects focus on creating new CRISPR systems that can specifically target and repair the NF1 gene without causing off-target effects. Despite these advancements, challenges remain, especially the risk of off-target effects where CRISPR/Cas9 could accidentally alter unintended genes.
RNA editing: RNA editing provides a way to restore protein function in mutant genes by correcting mRNA. Two main methods are spliceosome-mediated RNA trans-splicing (SMaRT) and engineered trans-splicing ribozymes. SMaRT leverages the cell’s splicing machinery and a pre-trans-splicing molecule (PTM) to modify target RNA by inserting new coding sequences, though non-targeted splicing remains a safety concern[11]. Engineered trans-splicing ribozymes specifically recognize and correct target RNA, replacing mutations with correct sequences. Once again, the size of the NF1 gene complicates the use of this technology. In addition, it is unclear whether the repair efficiencies of current trans-splicing ribozymes are medically realistic for the treatment of NF-1.
Antisense Oligonucleotides (ASOs): ASOs are synthetic molecules that modulate gene expression by binding to pre-mRNA, enabling inhibition of gene expression, exon inclusion, and exon skipping11. AOs can be conjugated to cell-penetrating peptides to improve uptake11. They can inhibit gene expression by targeting regulatory regions or causing exon skipping, which disrupts the reading frame and leads to mRNA degradation11. Leier et al.,15 investigates the feasibility of using exon skipping as a therapeutic approach for neurofibromatosis type I (NF1). The researchers focused on exon 17 of the NF1 gene, exploring whether its removal could maintain neurofibromin protein expression and function. The study found that skipping exon 17 restored functional neurofibromin expression in these cells. Additionally, a mouse model with a homozygous deletion of exon 17 showed viability and a normal lifespan, although some alterations in lymphoid organs were noted. This suggests that exon skipping could be a viable therapeutic strategy for NF1 patients, though further research is required to confirm its feasibility.
Nonsense suppression therapy (NST): Nonsense suppression therapy targets nonsense mutations, which introduce premature stop codons (PTCs) in mRNA, causing early termination of protein synthesis and mRNA degradation. These mutations account for about 11% of disease-causing mutations11. NST aims to bypass PTCs to produce full-length proteins and is being investigated for conditions like NF-1, where 20% of cases involve nonsense mutations. Potential treatments include suppressor tRNAs or drugs that induce PTC readthrough, potentially restoring enough protein function to reduce symptoms11.
Figure 1. Example of Potential Gene-Targeted Therapies for Neurofibromatosis Type 1.
Conclusion
Gene-targeted therapies represent a promising frontier in the treatment of Neurofibromatosis Type 1, offering potential solutions that address the genetic root of the disease rather than just its symptoms. Techniques such as gene replacement, RNA editing, and nonsense suppression therapy are at the forefront of this research. These approaches aim to restore or replace the function of the defective NF1 gene, thereby reducing tumor growth and improving patient outcomes. While challenges remain, particularly with the delivery and specificity of these therapies, ongoing research, and technological advancements continue to bring hope for more effective treatments.
The future of NF-1 treatment lies in the successful translation of these gene-targeted therapies from the laboratory to clinical practice. As our understanding of the genetic mechanisms underlying NF-1 deepens, and as new therapeutic strategies are developed and refined, there is potential for significant improvements in the quality of life for patients. Continued investment in research, coupled with clinical trials, will be crucial in overcoming the current limitations and realizing the full potential of these innovative treatments.
References
- Aronson J. Rare diseases, orphan drugs, and orphan diseases. BMJ: British Medical Journal 2006;333:127.
- Saleh M, Dib A, Beaini S, Saad C, Faraj S, El Joueid Y, et al. Neurofibromatosis type 1 system-based manifestations and treatments: a review. Neurological Sciences 2023;44:1931–47.
- Lee T-SJ, Chopra M, Kim RH, Parkin PC, Barnett-Tapia C. Incidence and prevalence of neurofibromatosis type 1 and 2: a systematic review and meta-analysis. Orphanet J Rare Dis 2023;18:292.
- Bayat M, Bayat A. Neurological manifestations of neurofibromatosis: a review. Neurological Sciences 2020;41:2685–90.
- Williams VC, Lucas J, Babcock MA, Gutmann DH, Korf B, Maria BL. Neurofibromatosis Type 1 Revisited. Pediatrics 2009;123:124–33.
- Friedman JM. Neurofibromatosis 1. GeneReviews 2022:[no pagination].
- Serra E, Puig S, Otero D, Gaona A, Kruyer H, Ars E, et al. Confirmation of a double-hit model for the NF1 gene in benign neurofibromas. Am J Hum Genet 1997;61:512.
- Gross AM, Wolters PL, Dombi E, Baldwin A, Whitcomb P, Fisher MJ, et al. Selumetinib in Children with Inoperable Plexiform Neurofibromas. New England Journal of Medicine 2020;382:1430–42.
- Adil A, Koritala T, Munakomi S, Singh AK. Neurofibromatosis Type 1 2023.
- Staedtke V, Anstett K, Bedwell D, Giovannini M, Keeling K, Kesterson R, et al. Gene-targeted therapy for neurofibromatosis and schwannomatosis: The path to clinical trials. Clinical Trials 2024;21:51–66.
- Leier A, Bedwell DM, Chen AT, Dickson G, Keeling KM, Kesterson RA, et al. Mutation-Directed Therapeutics for Neurofibromatosis Type I. Mol Ther Nucleic Acids 2020;20:739.
- Oluwapelumi Alao J, Joy Alao O, Precious Adebayo Z. Advancing Neurofibromatosis Type 1 Therapies through Basic Research Translation. International Research Journal of Oncology 2023;6:156–67.
- Heidenreich M, Zhang F. Applications of CRISPR–Cas systems in neuroscience. Nature Reviews Neuroscience 2015;17:36–44.
- Pickar-Oliver A, Gersbach CA. The next generation of CRISPR–Cas technologies and applications. Nature Reviews Molecular Cell Biology 2019;20:490–507.
- Leier A, Moore M, Liu H, Daniel M, Hyde AM, Messiaen L, et al. Targeted exon skipping of NF1 exon 17 as a therapeutic for neurofibromatosis type I. Mol Ther Nucleic Acids 2022;28:261–78.
About the Author
Carolina Ruivinho is an MSc research fellow at the RNA Systems Biology Lab, University of Lisbon, with a passion for science communication. She earned her BSc in Biology from the University of Coimbra in 2020 and completed her MSc with a thesis on miRNA-like sequences in the HIV genome. Post-MSc, she gained experience as a research fellow in both Portugal and Germany, consistently working with non-coding RNAs. Carolina is now focused on exploring RNA mechanisms to develop novel therapies, particularly using miRNAs.
Mentor: Dr.Margarida Gama Carvalho Affiliation: University of Lisboa