Genómica: conocimiento

En camino a comprender la arquitectura genética de los trastornos del espectro autista (TEA)

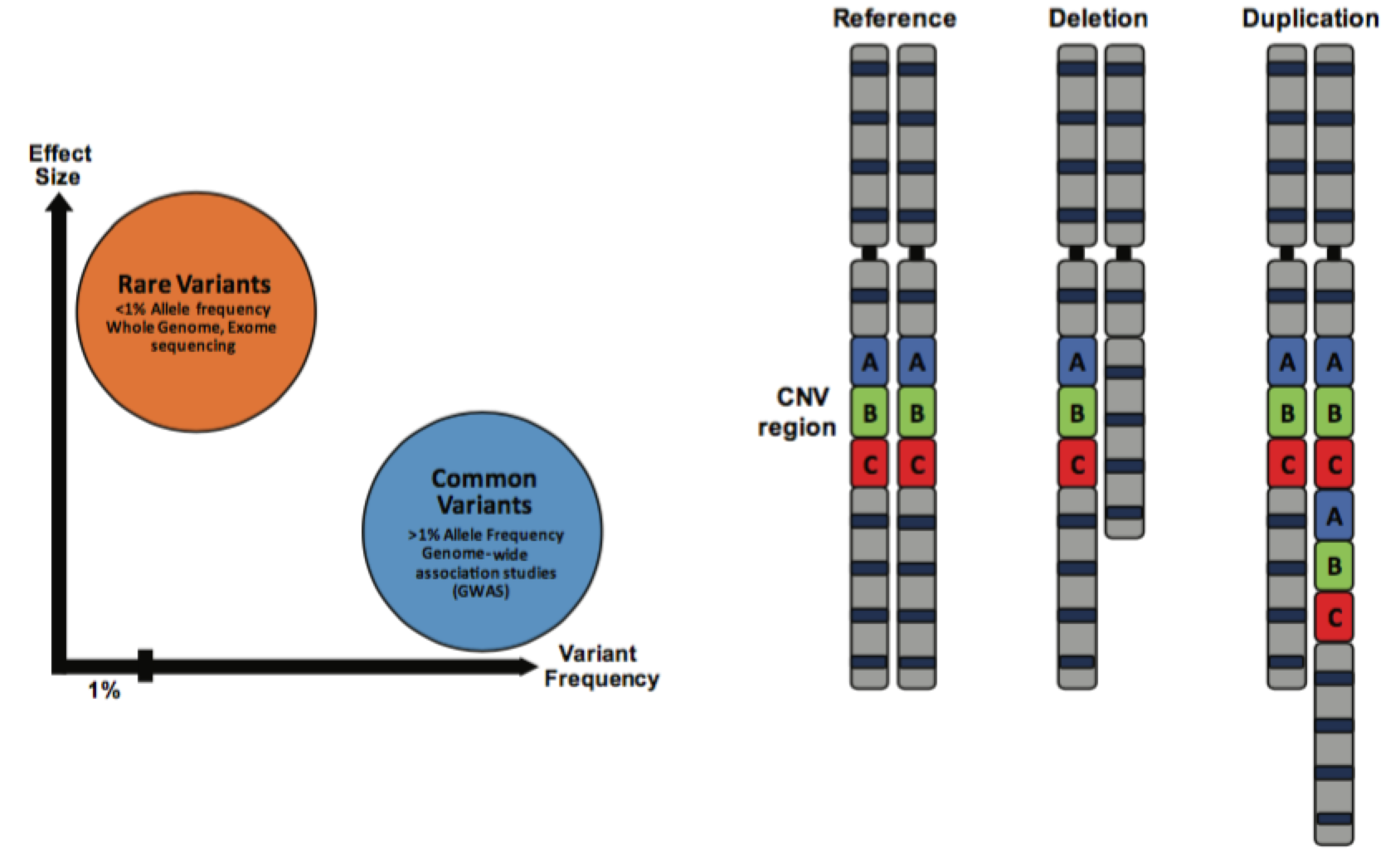
Los trastornos del espectro autista (TEA) son un grupo de trastornos del neurodesarrollo que se caracterizan por fenotipos cognitivos como intereses restringidos, conductas repetitivas y deficiencias en el lenguaje y la comunicación. Existe una enorme heterogeneidad fenotípica observada en los pacientes que se ve reflejada en un gran número de variantes genéticas identificadas para el TEA, sin que ninguna de ellas represente más del 1 % de los casos como máximo. Durante la década pasada se han hecho importantes avances en la identificación de variantes raras asociadas con el riesgo de TEA, y varios genes y loci de variantes de número de copias (Copy Number Variant, CNV) están ahora bajo observación para dilucidar los mecanismos moleculares asociados con la fisiopatología de TEA. Sin embargo, a pesar de que se sabe que la variación común contribuye en gran medida al riesgo de TEA[1], la búsqueda de variantes de riesgo comunes (polimorfismos de un solo nucleótido [single nucleotide polymorphisms, SNP]) asociadas al TEA no ha tenido tanto éxito, en gran parte debido a las limitaciones en el alcance estadístico de los estudios de asociación de genoma completo (genome-wide association study, GWAS) del TEA anteriores.
Un estudio reciente publicado en Nature Genetics finalmente aporta algunos datos sobre este asunto[2]. Al aumentar considerablemente el tamaño de la muestra en comparación con el análisis anterior, los autores pudieron identificar variantes de riesgo comunes únicas para el TEA. Además, el estudio demostró que el TEA comparte una cantidad sustancial de variantes de riesgo con otros trastornos psiquiátricos como la esquizofrenia (Schizophrenia, SCZ) y la depresión, algo que se planteó durante mucho tiempo como hipótesis debido a la alta comorbilidad observada entre estos trastornos.
"Se demostró una etiología compartida entre el TEA y otros trastornos psiquiátricos como la esquizofrenia (SCZ) y la depresión".
El estudio se basó en una cohorte de población danesa con más de 30,000 individuos, en la que un tercio de ellos eran casos de TEA[3]. Hubo registros clínicos completos disponibles para la cohorte, lo que permitió la separación en subgrupos con diferentes fenotipos cognitivos (TEA con o sin Discapacidad Intelectual [Intellectual Disability, ID], diagnóstico de síndrome de Asperger u otras comorbilidades, etc.). El análisis de esta cohorte identificó inicialmente marcadores significativos de todo el genoma en tres loci diferentes. A continuación, se obtuvieron datos de replicación utilizando cinco cohortes adicionales de ascendencia europea, y la ampliación del conjunto de datos con este lote adicional de pacientes permitió la identificación de nuevas variantes comunes de riesgo de TEA en dos loci adicionales.
Tras este descubrimiento inicial, los autores evaluaron el grado de correlación genética entre el TEA y otros rasgos cognitivos como la SCZ, la depresión y el éxito educativo. Se encontró una fuerte correlación, con diversos grados entre las diferentes categorías de TEA, y en comparación no se encontró ninguna correlación con otros rasgos como la altura o el índice de masa corporal, sin vínculos esperados con el TEA. Estos hallazgos permitieron profundizar en esos estudios de GWAS para descubrir nuevos loci que comparten el TEA y estos rasgos cognitivos, y se concluyó que había siete loci adicionales. En total, hay doce loci significativos en el genoma que están asociados sin lugar a dudas con el TEA.
La identificación de este grupo confiable de SNP permitió el cálculo de la heredabilidad del TEA dentro de diferentes categorías fenotípicas (es decir, pacientes con TEA con o sin ID). La heredabilidad es un concepto utilizado en genética de poblaciones que corresponde a la fracción de un rasgo dado que puede ser explicado por la genética. Entonces, por ejemplo, una enfermedad monogénica como la esclerosis tuberosa tendría una heredabilidad cercana al 100 % (si tiene la mutación específica, es casi seguro que tendrá la enfermedad), pero rasgos como la altura están mucho más influenciados por el entorno y, por lo tanto, su heredabilidad es de menor valor. Por lo tanto, se estimó que el TEA sin ID tenía una mayor heredabilidad que el que tenía ID. Curiosamente, dentro de los diferentes subgrupos de TEA, el síndrome de Asperger parecía tener casi el doble de heredabilidad que los demás.
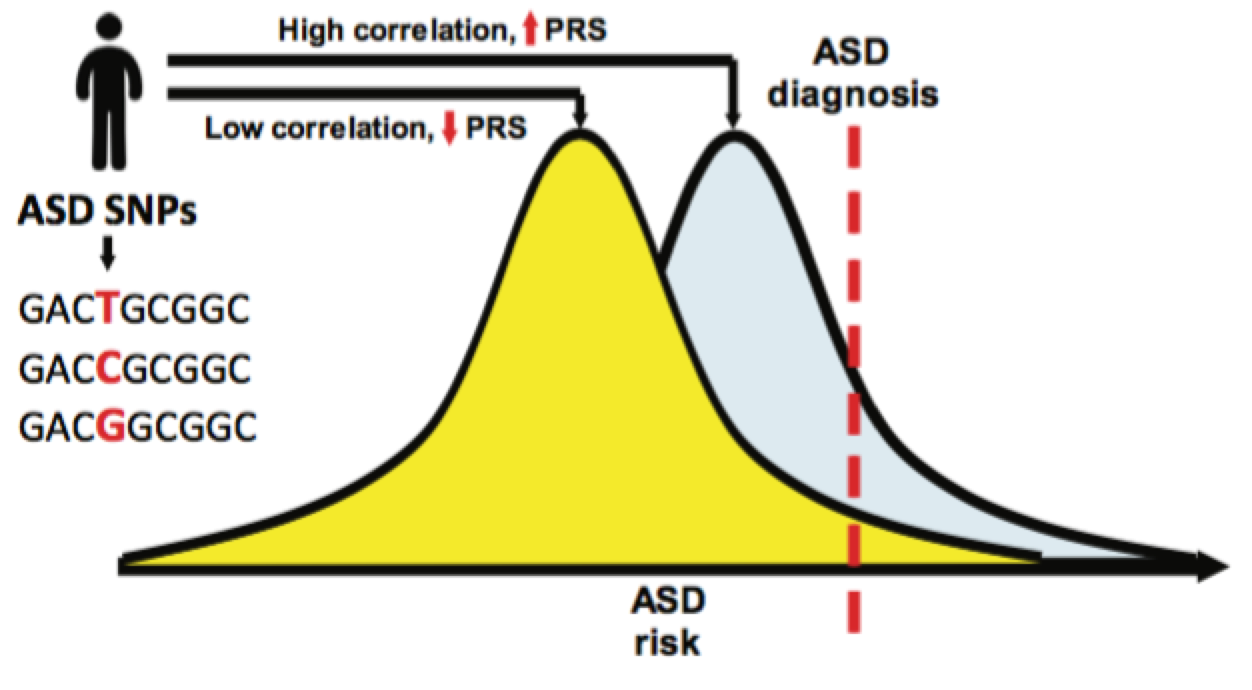
Más importante aún, la identificación de los SNP de TEA también permite el cálculo de su puntuación de riesgo poligénico (polygenic risk score, PRS). El PRS es una herramienta prometedora en el nuevo campo de la medicina personalizada y consiste en la estimación del riesgo en un paciente a partir de la correlación entre los SNP de los rasgos dados y las variantes genéticas del paciente. La comparación del PRS entre subgrupos de TEA y otras medidas cognitivas reveló una gran diferencia, lo que demuestra que la arquitectura genética entre los subgrupos de TEA es de hecho heterogénea.
La concentración funcional de los loci identificados mostró que están localizados en regiones de ADN conservadas y en secuencias potenciadoras, que son elementos reguladores que controlan la expresión de los genes. Un análisis adicional de estas marcas asociadas a potenciadores reveló que están concentradas en la corteza cerebral y en líneas celulares neuronales, especialmente en el cerebro en desarrollo. Además, los genes asociados a los SNP confiables de TEA estaban mayormente asociados a funciones neuronales y factores de transcripción, y se expresaban más durante la corticogénesis fetal. Estas observaciones son especialmente importantes ya que están de acuerdo con hallazgos previos sobre variaciones raras en TEA. Por ejemplo, la mutación rara más común de CNV encontrada en pacientes con TEA, la eliminación/duplicación del loci 16p11.2, se ha asociado con la desregulación de múltiples vías de señalización, específicamente en las neuronas corticales de la capa 3-4 durante el desarrollo temprano del cerebro[4].
La hipótesis más aceptada sobre la etiología del TEA hasta la fecha es que el TEA es un trastorno del desarrollo neurológico que comienza en las primeras etapas del desarrollo del cerebro. A pesar de la gran heterogeneidad genética observada con cientos de variantes de riesgo identificadas en docenas de genes o CNV diferentes, existe una convergencia en las vías funcionales más afectadas[5]. Muchos de los genes de riesgo asociados con el TEA, hasta la fecha, se incluyen generalmente en una de las siguientes categorías: asociados con funciones neuronales, que van desde la proliferación y diferenciación de progenitores neuronales hasta la formación y mantenimiento sinápticos, o asociados con la transcripción y remodelación de cromatina. Un ejemplo del primer grupo sería el SCN2A, un canal de sodio activado por voltaje responsable de la generación y propagación del potencial de acción en las neuronas, y un ejemplo del segundo grupo sería el CHD8, que regula la transcripción de varios genes.
"A pesar de la gran heterogeneidad genética observada (en el TEA), (…) existe una convergencia en las vías funcionales que se ven más afectadas".
La observación de que el primer estudio exitoso de GWAS sobre el TEA identifica un subconjunto de variantes que pueden asociarse con genes con propiedades similares a las identificadas anteriormente debería ser alentadora y emocionante para los científicos que trabajan en la caracterización de las vías moleculares afectadas. Además, el hecho de que hayamos alcanzado el umbral del tamaño de la muestra que permite que se tenga suficiente solidez estadística para la identificación de variantes allana el camino para nuevos análisis sobre diferentes subgrupos definidos, como por ejemplo si las personas responden a un determinado tratamiento. Este tipo de análisis se ha aplicado con éxito a otros trastornos psiquiátricos como la esquizofrenia[6].
En conjunto, la combinación de avances en los hallazgos genéticos y moleculares en la etiología del TEA seguramente favorecerá el descubrimiento y mejoramiento de los tratamientos y terapias. Una mejor comprensión de las causas genéticas del TEA, junto con los esfuerzos actuales en la medicina personalizada con base en pruebas genéticas, ayudará a los pacientes y a los médicos a encontrar el tratamiento más adecuado para cada caso. En combinación con los nuevos descubrimientos en la fisiopatología molecular del TEA que facilitan el descubrimiento de medicamentos, estamos por ver un gran cambio en el campo del TEA en los próximos años.
Referencias
[1]Gaugler, T., et al., Most genetic risk for autism resides with common variation. Nat Genet, 2014. 46(8): p. 881-5.
[2]Grove, J., et al., Identification of common genetic risk variants for autism spectrum disorder. Nat Genet, 2019. 51(3): p. 431-444.
[3]Pedersen, C.B., et al., The iPSYCH2012 case-cohort sample: new directions for unravelling genetic and environmental architectures of severe mental disorders. Mol Psychiatry, 2018. 23(1): p. 6-14.
[4]Lin, G.N., et al., Spatiotemporal 16p11.2 Protein Network Implicates Cortical Late Mid-Fetal Brain Development and KCTD13-Cul3-RhoA Pathway in Psychiatric Diseases. Neuron, 2015. 85(4): p. 742-54.
[5]Satterstrom, F.K., et al., Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. bioRxiv, 2019: p. 484113.
[6]Yu, H., et al., Five novel loci associated with antipsychotic treatment response in patients with schizophrenia: a genome-wide association study. Lancet Psychiatry, 2018. 5(4): p. 327-338.
Acerca del autor
Jorge Urresti es investigador postdoctoral en el Departamento de Psiquiatría de la Universidad de California, San Diego. Recibió su doctorado en Biología Molecular en la Universidad Autónoma de Barcelona. Su investigación se centra en dilucidar el impacto funcional de las mutaciones del TEA utilizando células derivadas de pacientes y en desarrollar aplicaciones de células madre para modelar trastornos del neurodesarrollo.