Genomics: Insight

A Deep Learning Approach to COVID-19 and Other Infectious Diseases
Predicting structural changes in viral proteins caused by genetic mutations
There are now nearly 3 million confirmed cases of COVID-19 worldwide, causing hundreds of thousands of deaths and economic pitfall. Recently, in a study that has yet to be peer-reviewed, researchers at China’s Zhejiang University stated that the coronavirus had mutated into at least 30 strains, and that pathogenicity differed between each strain1. This was presented as a possible explanation behind varying levels of severity of outbreaks around the world. As biotech companies and governments race towards developing a vaccine and treatments for the highly contagious disease, this finding raises the concern that certain treatments for one strain may not be effective for other strains due to the differences in protein structure, such as their receptors. Full or partial genomic sequences have been published for most known strains of COVID-19, but the gap in scientific knowledge currently lies in how these genetic mutations translate to physical protein structures. Without a concrete understanding of the associated proteins, vaccine and treatment development will be made much more difficult.
One solution to this problem can be found in machine learning. Machine learning is a branch of artificial intelligence that can discern patterns in data and use them to make predictions on new datasets. Researchers from DeepMind, an artificial intelligence research company based in the UK, recently published an article in Nature, detailing a new machine learning program named AlphaFold2. This system relies on the amino acid sequences, which are derived from the known DNA sequences, of proteins.
Background Information
Proteins are complex molecules that are essential to all biological organisms. DNA functions as a blueprint for building these structures, where an RNA transcript of three nucleotides of DNA encodes one specific amino acid. A chain of amino acids folds to form a protein. The way in which amino acid chains fold is extremely complex and difficult to predict. Currently, scientists can only go so far as to derive amino acid sequences from a line of genetic code. There is no other way to observe the intricate protein structure other than through time-consuming experimentation. For instance, 90% of the protein structures available in the Protein Data Bank were found through X-ray crystallography, which uses the density of electrons in three dimensions to infer the positions of atoms3. The main limitation preventing scientists from accurately predicting all protein structures is the lack of a generally agreed upon model to explain the complex nature of protein folding.
In the case of COVID-19, the protein folding problem comes into play in a very impactful way. As COVID-19 mutates into different strains, changes in the genetic code alter the amino acid sequence, which in turn, influence the final protein structure. Treatments for COVID-19 rely on fully understanding, for instance, the structure of the receptors on the surface of the virus, as a drug should be able to latch onto it in a lock and key mechanism and demobilize it. As such, there needs to be an efficient process by which complex protein structures can quickly be predicted based on the publication of the genetic sequences of new strains of coronavirus.
“As COVID-19 mutates into different strains, changes in the genetic code alter the amino acid sequence, which in turn, influence the final protein structure.”
Breakdown of the AlphaFold System
In their article, the DeepMind researchers explain that AlphaFold relies on deep neural networks. The DNNs take as input an amino acid sequence for a protein. Then, they output two predictions: the distance between amino acids and the angle of the chemical bonds between them. These outputs can then be used to produce a 3D diagram of the protein.
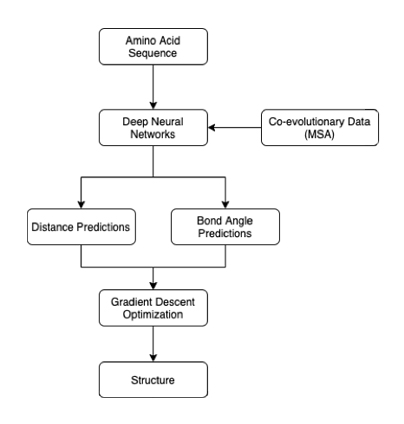
Figure 1: AlphaFold’s inner workings
AlphaFold is comprised of three neural networks; two find dimensional attributes of residues, while the third combines these predictions to solve for how close they are to the correct answer4. The system is dependent on co-evolutionary analysis. Multiple sequence alignment (MSA) highlights the similarities in amino acid sequences in homologous proteins. Similarities suggest that these fragments co-evolved over time, which then allows researchers to infer their close proximity in space. It is important to note that this approach is not to be confused with template-based modeling, as the researchers simply used data pertaining to similarities between sequences, instead of building off of an experimentally determined 3D model of another protein (the template).
However, the distances and bond angles predicted are not necessarily possible in a real protein. To solve this problem, the researchers initialize a protein structure that is empirically possible. Then, using a process known as gradient descent, the structure is iteratively refined to match the predicted distance and bond angles as closely as possible. In this way, generated structures can satisfy the predicted parameters, while still being physically possible. The model was trained on a dataset of 29,427 proteins from the Protein Data Bank.
In terms of performance, AlphaFold surpassed its counterparts. The Community Wide Experiment on the Critical Assessment of Techniques for Protein Structure Prediction, or CASP, is a biennial competition where researchers compete to computationally predict as many of the target proteins as accurately as possible. CASP consists of two categories: free modeling (FM) and template-based modeling (TBM). The free modeling category requires entries to predict the structure of a protein based on its sequence alone, while template-based modeling is the process of predicting an unknown protein’s structure based on proteins already in the Protein Data Bank.
Two metrics that are used by CASP to evaluate the new technique are the template modeling (TM) score and the global distance test total score (GDT_TS). Both calculate the similarity between two protein structures. The TM score measures how closely a predicted model matches the actual structure on a 0 to 1 scale. AlphaFold was able to predict 23 out of 43 free modeling domains with a TM-score above 0.7, while the next best entry achieved only 14 out of 43. More impressively, AlphaFold significantly increased the rate of progress from previous CASP competitions. The global distance test total score (GDT_TS) measures the gross topology of a prediction compared to the true structure on a scale of 0 to 100. AlphaFold roughly doubled the expected increase in GDT_TS from CASP12 to CASP135. CASP13 ranked entries based on summed, capped z-score, which essentially shows how far off from the average-performing model a model was–top entries have a positive summed z-score, while bottom entries receive a negative z-score. AlphaFold attained a summed z-score of 52.8 compared to the next best 36.6 in the FM category. In the TBM category, AlphaFold was able to perform on par with other models, despite not using any templates.

These developments in computational modeling of protein folding are very exciting. Although AlphaFold’s predictions are still not completely accurate, there is no doubt that it will have a considerable impact on the biomedical landscape. In the present, other researchers have expanded on the methods presented by DeepMind to create even more accurate models. Researchers at the Baker Lab published their findings on an upgraded version of AlphaFold named TrRosetta, which achieved an average TM score of 0.625 compared to AlphaFold’s 0.5876.
In future outbreaks of a highly contagious disease, AI-based protein structure prediction programs will help accelerate the discovery of vaccines and treatments, in addition to reducing costs. Experimentally determining protein structure remains expensive and time-intensive. For instance, X-ray crystallography requires costly equipment and takes several months to produce results. In contrast, AI-based protein modelling will yield near-instant results while only requiring DNA sequences and sufficient computing hardware, which is rapidly dropping in price. With AI tools such as AlphaFold, the time and cost from DNA sequencing to vaccine and drug development will significantly decrease.
“The cost of sequencing a genome continues to decrease, while the cost of experimentally determining protein structure remains expensive and time-intensive.”
About the Author
Simon Lee is a high school student at Whittle School and Studios in Washington, D.C. He is currently pursuing his interest in machine learning based solutions to global health problems.