Genomics: Insight

Diving Into the Cure for Cystic Fibrosis
Introduction
Cystic fibrosis is a genetic disorder that causes damage to internal organs, such as the lungs, because the condition negatively affects the cells that produce mucus. As a result, the mucus becomes thick, slimy, and viscous, rather than thin and slippery. Cystic fibrosis can also affect how the body produces sweat and digestive fluids.
Causes
Cystic fibrosis (CF) is caused by a mutation in the CFTR (cystic fibrosis transmembrane conductance regulator) gene. The mutation changes the protein that controls salts and fluids’ movement through the cells, affecting sweat, digestive fluids, and mucus. The severity of the disorder is dependent on which way the gene is mutated. In order to have CF, children must receive two mutated CFTR genes. With only one mutated gene, they will not have CF, but they could potentially pass it on to their children.
Symptoms
The symptoms vary significantly in terms of severity, but common symptoms include coughing, wheezing, and constipation. CF is progressive and can lead to further complications. Respiratory complications include damage in the lungs and airways. Cystic fibrosis can also lead to nutritional deficiencies, diabetes, and liver disease. Sometimes, CF reduces fertility in women and causes infertility in men. It can even cause osteoporosis, which is the thinning of bones.
Diagnosis
In the United States, all states test newborn children for Cystic Fibrosis using a blood test, DNA test, or sweat test. Blood tests examine the level of immunoreactive trypsinogen (IRT), a pancreatic enzyme, because people with CF have higher levels of IRT than normal. DNA tests are straightforward and look directly at the CFTR gene to locate any mutations. Sweat tests measure the amount of salt in a patient’s sweat, with higher levels indicating CF.
Current Solutions and Treatments
As of now, there is no way to heal CF permanently, but researchers are on the path to find a solution. However, there are ways to help alleviate the symptoms of CF through pills called modulators and through RNA therapy. Two methods that are currently being researched to cure CF for good are gene editing and gene therapy.
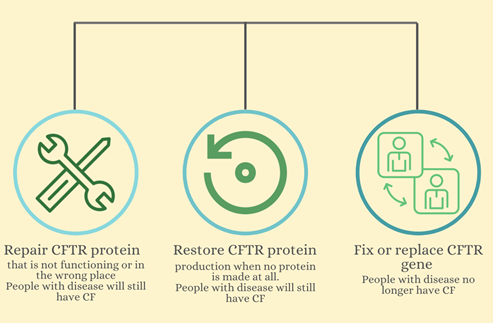 Figure 1: Three different approaches to relieve or cure CF. Modulators repair the CFTR protein, RNA therapy restores the protein, and gene editing and transfer could fix and replace the CFTR gene.
Figure 1: Three different approaches to relieve or cure CF. Modulators repair the CFTR protein, RNA therapy restores the protein, and gene editing and transfer could fix and replace the CFTR gene. Modulators
Doctors currently use CFTR modulators for various mutations to ensure the gene is performing correctly and in the right place. Despite having these modulators, there are only four different modulators that can repair a few mutations. As a result, the modulators are only useful in people with those specific mutations.
One modulator called Ivacaftor works by binding to the defective protein and opening the chloride channel at the cell’s surface. As a result, the gateway opens, allowing chloride to pass through, and the cell can regulate the movement of fluids in and out of the cell. Modulators that do this are called potentiators, and they can help with gating mutations, residual function mutations, splicing mutations, and conduction mutations. These defects alter the effectiveness and number of the proteins produced and the chloride’s access through the CFTR channel.
People who have an F508del mutation, the most common type of mutation, in both CFTR genes, can use the Lumacaftor/Ivacaftor modulator. Just like how Ivacaftor is known as a potentiator, Lumacaftor is a corrector modulator. It forms the F508del protein into the right shape and helps the protein stay at the cell’s surface for longer. With Lumacaftor alone, about a third of the sufficient chloride ions can pass through. The Lumacaftor/Ivacaftor combination holds the gate open, allowing the necessary amount of chloride to pass through the cell membrane.
Tezacaftor/Ivacaftor is another corrector and potentiator modulator combination that works similarly to the Lumacaftor/Ivacaftor one. However, it has fewer side effects and works not only with the double F508del mutation, but 26 other single-gene mutations as well.
Elexacaftor/Tezacaftor/Ivacaftor (Trikafta) adds Elexacaftor, another corrector, to the Tezacaftor/Ivacaftor modulator. Since Trifkafta is a combination of three others, it is known as “the triple combo.” Elexacaftor corrects another defect in the F508del protein’s formation and works for a larger number of CF patients who have different mutations. This modulator can be used for people who have the F508del mutation in one CFTR gene and any mutation in the other CFTR gene.
RNA Therapy:
RNA therapy is another way to relieve symptoms of CF. RNA is a messenger copy of DNA sent to the ribosomes as an instruction to create proteins. In patients with CF, since the DNA sequence of the CFTR gene is mutated, the mRNA is mutated, resulting in dysfunctional, few, or no proteins produced at all. RNA therapy would restore the mRNA sent to the ribosomes to ensure enough functional CFTR proteins are produced. However, RNA therapy is still being developed and is not used to treat CF yet.
Gene Editing/Gene Therapy:
Gene editing and gene therapy could potentially remove the disease from a patient’s body by either fixing or replacing the gene. Each organism has its own way of repairing any damaged DNA, as DNA is vital for cells to function. Gene editing utilizes a repairing mechanism from bacteria to fix the CFTR gene with a correct sequence that genetic engineers design. Gene therapy is similar, but it places a transgene with the correct CFTR gene somewhere else in the cell or DNA, other than where it normally is. There are also various limitations to the duration of the transgene inside the cell (Figure 2). However, inserting either mechanism inside all of the trillions of cells in a human to correct the mutation is difficult.
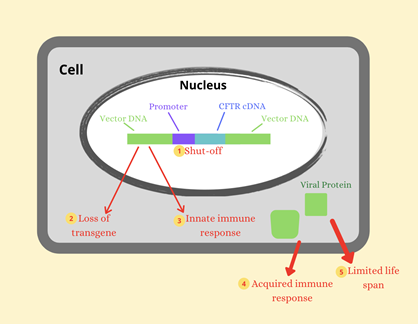 Figure 2: Factors limiting the duration of transgene expression in airway surface tissue.
Figure 2: Factors limiting the duration of transgene expression in airway surface tissue.As these methods continue to be researched every day, scientists come closer and closer to a permanent cure for Cystic Fibrosis, saving the lives of many.
About the Author
Leisha Devisetti is a 15-year-old high school sophomore who attends The Harker School in San Jose, California. She has a passion for science and loves to learn how researchers find cures by testing diseases or viruses that seem incurable.